
LEUCEMIA LINFATICA CRONICA
La leucemia linfatica cronica (LLC) è un tumore del sangue causato dall’accumulo, a livello di sangue periferico, midollo osseo, linfonodi e milza, di un particolare tipo di cellule del sistema immunitario, i linfociti B maturi. In questa forma di leucemia, i linfociti si accumulano a seguito di una mutazione che li porta a formare un clone, ovvero a replicare cellule tutte uguali fra di loro e con delle proprietà di sopravvivenza superiori alle cellule normali. Si tratta di una malattia dinamica, che trae la sua origine da una ricca serie di eventi biologici e genetici primari e secondari. L’interazione con gli antigeni, lo stato di “attivazione” cellulare che ne segue, e le complesse interazioni con il microambiente plasmano una malattia dal decorso eterogeneo, talora preceduta da una condizione predisponente, la linfocitosi B-monoclonale (monoclonal B-cell lymphocytosis, MBL).
La malattia si definisce cronica perché, solitamente, la velocità di replicazione di queste cellule è bassa e i sintomi, se presenti, si sviluppano lentamente.
L’andamento clinico può tuttavia essere molto diverso; in alcuni casi la linfocitosi è di entità molto modesta e si mostra stabile nel tempo, in altri casi invece può crescere più rapidamente. La causa della LLC non è ben chiara, ma è noto come alcune alterazioni genetiche modifichino i meccanismi di proliferazione cellulare contribuendo alla patogenesi della malattia. Venendo a mancare un controllo efficace sulla loro produzione e sopravvivenza, il numero dei linfociti tende a crescere. L’entità della malattia e la sua evolutività dipendono da quali e quanti meccanismi di regolazione sono compromessi. L’identificazione delle caratteristiche genetiche è utile, nei pazienti che richiedono una cura, anche per indirizzare la scelta dei farmaci da impiegare.
INCIDENZA
La LLC è la forma di leucemia più frequentemente osservata nei paesi occidentali dove rappresenta circa il 25-30% di tutte le leucemie dell’adulto, mentre è molto più rara nei paesi asiatici. Questa forma di leucemia interessa soprattutto i soggetti di sesso maschile. L’età media alla diagnosi è attorno ai 70 anni, e l’incidenza aumenta da 1 caso/anno/100.000 abitanti nella fascia 40-50 anni a 20 casi nella fascia 70-80 anni. Oltre il 40% delle LLC è diagnosticata ad un’età >75 anni, mentre meno del 10% è diagnosticata prima dei 50 anni
Pur non essendo una malattia ereditaria o trasmissibile, sono note alcune famiglie in cui sono presenti più casi di Leucemia Linfatica Cronica.
CAUSE
I linfociti sono cellule del sistema immunitario responsabili della difesa dell’organismo da agenti potenzialmente pericolosi. Quando incontrano il loro bersaglio, i linfociti si attivano e cominciano a moltiplicarsi, in modo da creare un numero di cellule sufficiente ad affrontare l’infezione e, una volta che quest’ultima sia stata risolta, la maggior parte dei linfociti attivati, ormai inutili, muore.
Nella leucemia linfatica cronica uno dei linfociti attivati continua a moltiplicarsi anche quando non è più necessario, dando vita a un numero sempre maggiore di cellule, che costituiscono la leucemia.
Nella maggior parte dei casi, le cellule della malattia presentano delle alterazioni a carico del materiale genetico (DNA). Queste alterazioni colpiscono spesso geni importanti per il controllo della crescita dei linfociti, e contribuiscono alla loro trasformazione in cellule leucemiche. Tuttavia, si pensa che anche altri fattori, come la stimolazione del sistema immunitario da parte di agenti estranei, siano coinvolti nello sviluppo della malattia.
I fattori genetici possono predisporre allo sviluppo della malattia: circa il 10% dei pazienti hanno dei familiari affetti da leucemia linfatica cronica o da altre malattie del sistema linfatico. Invece i fattori ambientali come le radiazioni e le sostanze chimiche sembrano essere meno importanti, in quanto non aumentano il rischio di sviluppare la malattia.
CLASSIFICAZIONE
I linfociti presenti nel nostro corpo sono di due tipi diversi: B e T. Nella grande maggioranza dei casi (il 95%), a dare origine alla malattia è un linfocita B. La leucemia linfatica cronica di tipo B può essere classificata in base alle caratteristiche molecolari delle cellule leucemiche (es. alterazioni dei cromosomi, mutazioni a carico di alcuni geni o espressione di alcune proteine). Alcune di queste caratteristiche molecolari sono associate a una malattia più aggressiva, o più resistente ai farmaci. Per questo la classificazione molecolare può aiutare a prevedere l’andamento clinico dei pazienti e a individuare il trattamento più opportuno.
Le alterazioni più frequenti nelle cellule tumorali dei pazienti affetti di leucemia linfatica cronica sono: – perdita di una porzione del cromosoma 13 (del13q14), presente in circa il 55% dei pazienti e associata, in assenza di altre anomalie cromosomiche, a un decorso della malattia favorevole;
– perdita di una porzione del cromosoma 11 (del11q23);
– acquisizione di una terza copia del cromosoma 12 (trisomia 12);
– perdita di una porzione del cromosoma 17 (del17p13), associata allo sviluppo di resistenza al trattamento, e quindi a un decorso clinico sfavorevole;
– mutazioni nel gene TP53, che hanno effetti simili a quelli della perdita del cromosoma 17.
Tali analisi approfondite vengono solitamente effettuate solo nei pazienti che necessitano di essere trattati, e non in quelli asintomatici.
SINTOMI E SEGNI
La maggior parte delle persone con LLC non ha alcun sintomo e si accorge di questa malattia solo nel corso di analisi di controllo che rivelano un aumento dei globuli bianchi. La malattia viene quindi spesso diagnosticata in modo del tutto casuale, in persone che stanno bene e che hanno eseguito delle analisi del sangue per altri motivi. Nelle persone che hanno sviluppato una malattia più attiva, la LLC può manifestarsi con un aumento della grandezza dei linfonodi, che diventano palpabili, talvolta in più sedi (collo, ascelle, inguine, ecc.). I linfonodi aumentati di volume in sedi “profonde”, come quelli del torace e dell’addome, sono identificabili solo con esami ecografici e radiografici. Molto raramente, possono esservi alcuni sintomi come il dimagramento e la sudorazione notturna che si osservano solitamente in altre malattie come i linfomi. In alcuni casi, la LLC viene diagnosticata in persone che hanno una infezione.
Figura 1. Sintomi della malattia LLC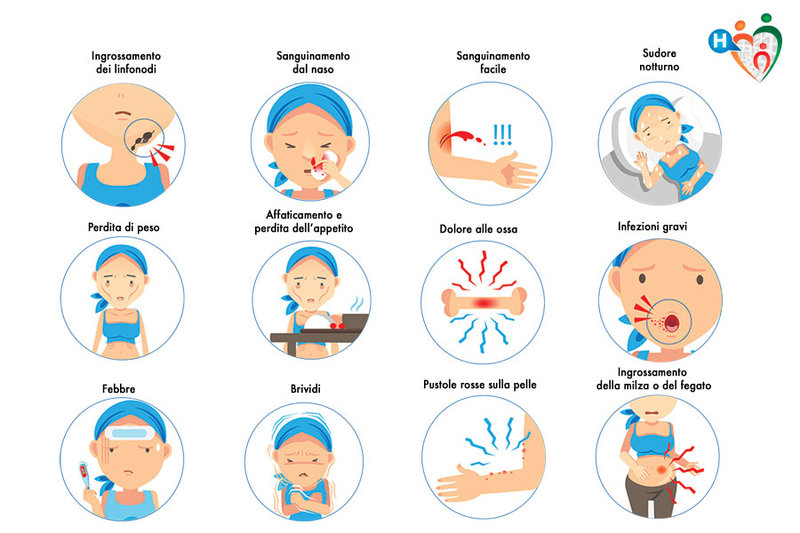
DIAGNOSI ED ESAMI
Il primo esame da fare per identificare una leucemia linfatica cronica è l’emocromo, che permette di misurare la quantità dei diversi tipi di cellule presenti nel sangue: in caso di leucemia linfatica cronica, il numero dei linfociti risulta aumentato e il rapporto tra le varie popolazioni di globuli bianchi è anormale (inversione della formula leucocitaria). È importante, tuttavia, considerare il numero assoluto dei linfociti e non le alterazioni percentuali degli stessi.
Figura 2. Striscio di sangue periferico

Tuttavia, la quantità di linfociti nel sangue può aumentare anche a causa di altre malattie (come le infezioni virali). Per escludere questa possibilità, si effettua un’analisi immunofenotipica, che permette di esaminare le caratteristiche di superficie dei linfociti e di riconoscere quali di questi siano leucemici. L’analisi immunofenotipica consente anche di misurare con grande precisione il numero di cellule leucemiche: infatti per poter diagnosticare una leucemia linfatica cronica, il sangue del paziente deve contenere almeno 5 milioni di linfociti per millilitro (o 5.000/μL), per almeno tre mesi di seguito (in caso di infezioni, invece, l’aumento dei linfociti è di minore durata). Nel caso in cui si riscontrino linfociti monoclonali < 5.000/ μL, si pone diagnosi di linfocitosi B-monoclonale (MBL), una condizione che può predisporre allo sviluppo della malattia. Una volta stabilita la diagnosi è necessario definire lo stadio clinico, che si associa ad un diverso andamento clinico e determina la necessità o meno di iniziare un trattamento.
In alcuni casi è necessario anche effettuare esami radiologici, analisi del midollo osseo (che viene prelevato in anestesia locale dalle ossa del bacino) o biopsie (cioè prelievi di tessuto) dei linfonodi.
STADIAZIONE DI MALATTIA
Una volta che la diagnosi di LLC è stata completata, viene valutato lo stadio della malattia, cioè quelle caratteristiche che meglio definiscono l’entità/estensione della malattia. Vengono utilizzati 2 sistemi di stadiazione: quello proposto a Rai (5 stadi: da 0 a IV) e quello proposto da Binet (3 stadi: A-B-C), che sono tra loro largamente sovrapponibili.
Lo stadio viene definito in base alla presenza o meno dell’aumento del volume della milza, del fegato e dei linfonodi e in rapporto al valore di emoglobina e al numero di piastrine.
Nel midollo osseo vengono prodotti i globuli rossi, le piastrine e altri tipi di globuli bianchi, come i granulociti. Se la presenza dei linfociti della LLC nel midollo è molto importante, questa può determinare una ridotta attività del midollo osseo, con conseguente riduzione del numero di globuli rossi (anemia) e del numero delle piastrine (piastrinopenia).
Una cura della LLC viene iniziata quando lo stadio è avanzato o quando la malattia mostra chiari segni di rapida e significativa crescita.
DECORSO DELLA MALATTIA
Il decorso della LLC è molto variabile da persona a persona.
In base alla velocità di progressione della malattia, si possono distinguere casi più lenti nella evoluzione, che necessitano di trattamenti anche a distanza di un decennio dalla diagnosi iniziale. Altri pazienti invece manifestano una malattia più aggressiva, che porta alla necessità di trattamenti dopo pochi mesi o anni dalla diagnosi. Le due forme di malattia sono in molti casi distinte dalla presenza o assenza di caratteristiche biologiche tra cui difetti genetici a carico del gene TP53 o mutazioni nei geni delle immunoglobuline (IGHV). Di solito queste mutazioni non sono ereditarie, ma acquisite, e sono infatti presenti soltanto nelle cellule tumorali.
Circa il 70-80% dei pazienti alla diagnosi di LLC non richiede trattamento, ma sarà sottoposto a monitoraggio periodico presso il MMG e/o presso l’ambulatorio specialistico.
Il trattamento sarà iniziato se e quanto saranno presenti i criteri di progressione di malattia: MALATTIA ATTIVA
La malattia si definisce ATTIVA quando vi sia almeno una delle seguenti condizioni:
1. Progressiva citopenia (anemia e/o piastrinopenia) da infiltrazione midollare di malattia
2. Splenomegalia massiva (almeno 6 cm dal margine costale) o progressiva o sintomatica
3. Massive linfoadenopatie (almeno 10 cm di diametro massimo) o adenopatie progressive o sintomatiche
4. Linfocitosi progressiva con un incremento dei linfociti > 50% in due mesi o un raddoppiamento dei linfociti (LTD) in un tempo inferiore ai 6 mesi in pazienti con una conta linfocitaria iniziale > 30.000/μl.
5. Anemia o piastrinopenia autoimmune non responsive al trattamento steroideo o ad altre
terapie standard
6. Coinvolgimento extranodale sintomatico o funzionale (cute, rene, polmone, colonna)
7. Sintomi sistemici definiti dalla presenza di almeno una delle seguenti condizioni:
▪ calo ponderale ≥10% nei 6 mesi precedenti;
▪ ECOG (Eastern Cooperative Oncology Group Performance status) PS >2;
▪ febbre per più di 2 settimane senza evidenza di infezione
▪ sudorazioni notturne per più di un mese senza evidenza di infezioni.
Il valore assoluto della linfocitosi, indipendentemente dalla conta linfocitaria, o la linfocitosi progressiva, in assenza di altre indicazioni, non rappresentano di per sé indicazione all’inizio del trattamento.
Nemmeno la presenza di una o più componenti monoclonali, l’ipogammaglobulinemia, né la diatesi infettiva.
TERAPIA
Per quanto riguarda il trattamento, esistono criteri internazionalmente riconosciuti per l’avvio della terapia.
Nei pazienti che non presentano sintomi o criteri di trattamento, la malattia deve essere semplicemente monitorata nel tempo (“watch and wait”), poiché non vi è vantaggio a iniziare una terapia precocemente.
Nei pazienti sintomatici o che hanno raggiunto i criteri specifici di trattamento, invece, la terapia viene scelta dal medico in base alle caratteristiche cliniche della malattia, all’età e allo stato di salute del paziente.
I fattori biologici (i più importanti sono: – la perdita di una parte del cromosoma 17; – la presenza di mutazioni dei geni TP53; – lo stato mutazionale di IGHV), che hanno una valenza prognostica (in quanto predicono l’andamento della malattia) e predittiva (in quanto predicono la risposta al trattamento), possono essere valutati mediante esami molecolari sul sangue periferico o, raramente, sul midollo osseo.
Analogamente alla biopsia ossea e alla TC, tali esami sono consigliabili nel momento in cui c’è la necessità di iniziare una terapia.
La scelta della terapia di prima linea, che mira a migliorare la qualità di vita e aumentare la sopravvivenza, si basa su caratteristiche della malattia, su caratteristiche del paziente (età, comorbidità e trattamenti in atto, stato di salute generale), sulla preferenza del paziente e/o sulla presenza di un caregiver.
Negli ultimi anni si è molto modificato l’approccio terapeutico a questa patologia; infatti, la chemioterapia è stata completamente sostituita da farmaci biologici ed anticorpi monoclonali (questi ultimi già utilizzati in precedenza in associazione alla chemioterapia).
ANTICORPI MONOCLONALI
Gli anticorpi monoclonali sono proteine anticorpali che riconoscono e “lisano” i linfociti della LLC. Quelli più largamente impiegati nella LLC sono il rituximab e l’obinutuzumab (diretti verso l’antigene CD20). Le prime somministrazioni degli anticorpi monoclonali possono essere seguite da reazioni “allergiche” che solitamente si attenuano o scompaiono con le somministrazioni successive. Per prevenire le reazioni infusionali agli anticorpi monoclonali vengono somministrati alcuni farmaci (“premedicazione”: cortisonici, anti-istaminici e paracetamolo).
I FARMACI “BIOLOGICI”
Introdotti per il trattamento della LLC alcuni farmaci “biologici”, orali: ibrutinib, acalabrutinib, zanubrutinib, idelalisib, venetoclax. Si tratta di agenti che interferiscono sui meccanismi responsabili della crescita e sopravvivenza delle cellule leucemiche, concentrandosi su anomalie specifiche presenti all’interno delle cellule tumorali. Bloccando queste anomalie possono causare la morte delle cellule tumorali. Tra queste opzioni terapeutiche si annoverano gli inibitori di una chinasi intracellulare chiamata tirosin chinasi di Bruton (BTK) ovvero ibrutinib, acalabrutinib e zanubrutinib, e gli inibitori di una molecola intracellulare che impedisce la morte cellulare chiamata Bcl2 (il farmaco si chiama venetoclax); e, ormai di minore impiego clinico per la significativa tossicità, gli inibitori di fosfatidil-inositolo-3-chinasi-delta (PIK3D), ovvero idelalisib.
Questi farmaci, all’inizio impiegati come terapia continuativa fino a progressione o a tossicità che rendesse necessaria la sospensione del trattamento stesso, possono essere oggi impiegati anche in combinazione, con lo scopo di poter trattare il paziente per un periodo limitato di tempo (12, 15, 24 mesi), garantendogli un periodo più o meno lungo di “vacanza terapeutica” (un periodo libero da trattamento in cui il paziente esegue solo controlli clinici e di laboratorio, senza assumere alcun farmaco specifico per la malattia).
Al termine del trattamento la malattia viene valutata attraverso esami strumentali e analisi del midollo osseo. In presenza di risposta al trattamento il paziente torna in una fase di follow-up (watch and wait).
I nuovi farmaci biologici rappresentano oggi la terapia di prima linea di scelta per la maggior parte dei pazienti, ai quali si possono offrire terapie orali con inibitori di BTK in via continuativa, vale a dire sino a progressione o a sviluppo di tossicità significative, ovvero una terapia di durata definita (12-15 mesi) utilizzando l’inibitore di BCL2 venetoclax in combinazione con l’anticorpo monoclonale anti CD20 obinutuzumab (terapia infusionale che richiede accesso in Day Hospital), o in combinazione con ibrutinib.
I farmaci disponibili, seppur molto efficaci nel tenere sotto controllo la malattia per periodi prolungati, anche di anni, non sono oggi in grado di guarire i pazienti.
Al momento della ricaduta, attesa nella maggior parte dei casi, si possono impiegare successivamente e in modo sequenziale tutti i trattamenti che non sono stati utilizzati in prima linea, aggiungendo ulteriori anni di controllo della malattia e dei sintomi correlati.
I nuovi farmaci biologici, agendo su un bersaglio preciso sono più efficaci e meglio tollerati rispetto alla tradizionale chemioterapia, e per questo hanno migliorato molto la prognosi dei pazienti con leucemia linfatica cronica.
Tuttavia, queste molecole hanno specifiche tossicità che rendono necessaria un’attenta valutazione del paziente e delle sue patologie di base, per la migliore scelta terapeutica, che oggi è sempre più “ritagliata sul paziente” e tiene conto, oltre che delle caratteristiche biologiche della malattia, anche delle comorbidità.
Pertanto, per l’avvio di un trattamento ottimale, è importante considerare:
1. Fattori correlati alla malattia: caratteristiche biologiche della malattia (TP53 o del 17p, IGHV)
2. Fattori correlati al paziente e alla scelta del migliore farmaco:
a. Età e condizioni generali (Fitness status)
b. Patologie preesistenti (Comorbidità)
c. Terapie concomitanti (per il rischio di interazioni)
d. Preferenza del paziente (terapia limitata nel tempo vs terapia a lungo termine)
e. Via di somministrazione del farmaco e compliance del paziente (terapia endovenosa vs terapia assunta per bocca)
Trial clinici: è possibile che il medico ematologo proponga al paziente l’arruolamento all’interno di uno studio sperimentale. Gli studi sperimentali sono delle grandi occasioni in quanto viene offerto al paziente un farmaco o una combinazione di farmaci non ancora in commercio, ma del quale il paziente potrebbe beneficiare in ragione della partecipazione allo studio stesso.
IL TRAPIANTO ALLOGENICO DI CELLULE STAMINALI
Attualmente, l’unico approccio terapeutico potenzialmente in grado realmente di eradicare e quindi “guarire” la LLC è il trapianto allogenico di cellule staminali emopoietiche. La fattibilità di questo approccio terapeutico è limitata da diversi fattori. Innanzitutto, la LLC è una forma di leucemia che interessa le persone meno giovani che hanno un’età non adeguata a tollerare i trattamenti che sono impiegati nella procedura trapiantologica. Inoltre, su questa procedura gravano ancora complicazioni che possono essere più pronunciate nel soggetto anziano. Infine, deve esservi la disponibilità di un donatore compatibile.
Oggi il trapianto allogenico (da familiare o da donatore) di cellule staminali ematopoietiche è indicato in casi selezionati, tra cui pazienti giovani con recidiva di leucemia linfatica cronica resistente a tutti i nuovi farmaci biologici e con caratteristiche biologiche sfavorevoli. Può anche essere preso in considerazione per pazienti in cui la malattia è progredita in un linfoma aggressivo (sindrome di Richter).
LE COMPLICAZIONI IN CORSO DI LLC
LE INFEZIONI
I pazienti con LLC, soprattutto se hanno già eseguito molti trattamenti, possono presentare difese immunitarie meno efficaci. La maggiore vulnerabilità alle infezioni può essere più accentuata se si associa anche alla riduzione dei granulociti e degli anticorpi indotta dalle cure. Tra le infezioni più frequenti vanno ricordate quelle polmonari, le infezioni virali erpetiche, la varicella, l’herpes varicella-zoster (fuoco di S. Antonio).
- LE VACCINAZIONI
La vaccinazione contro lo pneumococco, il germe frequentemente in causa nelle polmoniti, e quella stagionale antinfluenzale possono essere utili nel ridurre il rischio infettivo. La vaccinazione antinfluenzale con virus inattivati e il vaccino anti-Covid a mRNA sono consigliati a tutti i pazienti e ai loro familiari. In corso di terapia la risposta immunitaria è meno efficace; quindi, il vaccino può non proteggere dall’infezione, ma è comunque raccomandato.
- LE IMMUNOGLOBULINE
In presenza di infezioni polmonari ricorrenti e ipogammaglobulinemia, potrebbe essere raccomandata regolare infusione di immunoglobuline.
ANEMIA EMOLITICA AUTOIMMUNE
La minore competenza immunitaria può condizionare, specie in pazienti con malattia più avanzata, la comparsa di disordini autoimmuni, in particolare, la comparsa di un’anemia associata a ittero (colore giallastro della cute) dovuta alla presenza di anticorpi patologici diretti contro i propri globuli rossi.
ALTRI TUMORI
Le persone che hanno una LLC possono avere difese immunitarie che li rendono un po’ più fragili anche per lo sviluppo di altri tumori. E’ importante quindi evitare fattori di rischio aggiuntivi quali il fumo.
Tutti gli esami necessari per una adeguata prevenzione devono essere eseguiti periodicamente (visite ginecologiche, Pap test, ecografia mammaria, mammografia, PSA, visita urologica, sangue occulto fecale, ecc).
TRASFORMAZIONE IN RICHTER
La trasformazione in Richter è una complicazione rara che interessa persone con malattia solitamente avanzata e sottoposta a molteplici trattamenti. Questa condizione è dovuta all’emergenza di una malattia più grave. Si sviluppa un linfoma, nella maggior parte dei casi a partenza dai linfociti stessi della LLC, che acquisiscono caratteristiche di maggiore aggressività. La sindrome di Richter richiede un trattamento diverso da quello della LLC. È quindi importante che la sindrome di Richter sia riconosciuta e trattata appropriatamente. Qualora vi sia il sospetto della comparsa di una sindrome di Richter sono richieste indagini radiologiche (TAC PET) e la biopsia PET guidata di un linfonodo e del midollo osseo.
VIVERE CON UNA LEUCEMIA LINFATICA CRONICA
Nella maggior parte dei casi, la diagnosi di leucemia linfatica cronica non ha conseguenze immediate sulla vita del paziente. Se la malattia è stabile e asintomatica, infatti, il paziente deve presentarsi in ospedale solo per visite di controllo semestrali o annuali, durante le quali si sottopone a un esame medico e alle analisi di routine.
La malattia può mantenersi stabile anche per anni; solo in caso di progressione si procede con il trattamento. Dal momento che le terapie attuali non permettono una guarigione, la ricaduta dopo il trattamento è attesa; tuttavia, in molti pazienti è possibile mantenere la malattia sotto controllo per molti anni.
Alla luce di quanto detto, nel paziente con leucemia linfatica cronica è importante:
- Considerare profilassi vaccinali:
• Alla diagnosi raccomandate: Pneumococco (PCV20), Herpes Zoster (aRZV), (E’ possibile che al paziente inviato al Centro Vaccinale vengano proposte anche vaccinazioni anti Haemophilus e Meningococco)
• Annuali: Influenza, SARS-CoV-2
- Effettuare screening oncologici raccomandati per età e sesso, anche per tumori cutanei non melanoma.
Dott.ssa Sara Marinoni
UOC Ematologia, Ospedale di Legnano, ASST Ovest Milanese
Gli ultimi aggiornamenti scientifici:
Leucemia Mieloide Cronica: terapia e qualità di vita
Globalmente la Leucemia Mieloide Cronica (LMC) rende ragione del 15-20% di tutti i casi di leucemia diagnosticati in età adulta. In particolare in letteratura viene riportata un’incidenza annua di circa 1.1-1.8 nuovi casi ogni 100.000 persone, con un’età mediana al...

I successi del progetto di genomica finanziato da AIL Milano
Tra il 2014 e il 2017, AIL Milano ha finanziato un importante progetto di ricerca scientifica, con un contributo complessivo di 438 mila euro. Lo studio è stato realizzato presso l’Istituto Nazionale dei Tumori di Milano, coordinatore scientifico il Prof. Paolo...

L’evoluzione della medicina nella cura delle malattie onco-ematologiche
Prof. Paolo Corradini Direttore del Dipartimento di Ematologia e Onco-Ematologia Pediatrica, Fondazione IRCCS Istituto Nazionale dei Tumori Università degli Studi di Milano AIL Milano nasceva 40 anni fa. Quale era la situazione della ricerca scientifica sui...

Dalla parte del paziente: Educare il sistema immunitario ad attaccare il tumore
In questo articolo della nostra rubrica vogliamo informarvi riguardo ad una delle maggiori innovazioni degli ultimi anni nel campo della terapia antitumorale. L'argomento che tratteremo è l'utilizzo del nostro stesso sistema immunitario come difesa contro il tumore....















